
An estimated 128,000 hospitalized patients die each year from Adverse Drug Reactions (ADRs. aka Adverse Events, AEs), which matches stroke as the 4th leading cause of hospital deaths (see here). Deaths and serious reactions outside of hospitals would signicantly increase the totals. This does not include deaths and hospitalizations from over-dosing, errors, or recreational drug use. We know this because of hundreds of thousands of drug “Adverse Event Reports” (AERs) received by the FDA every year directly from healthcare professionals (HCPs such as physicians, pharmacists, nurses and others), consumers (such as patients, family members, lawyers and others), and drug companies, which are normally required to send AERs it receives from HCPs to the FDA.
The following chart shows the trend in AERs received by the FDA from 2004 through 2015.

I have analyzed data from 2003 through 2014 to look at the number of AERs submitted by HCPs versus consumers, the number of serious adverse events versus the number of adverse events involving death, and the correlation between serious AEs and user fees paid to the FDA by drug companies. I see some interesting trends in the data.
In 2014 – for the first time – more AERs were submitted by consumers than by HCPs as shown in the following two chart.
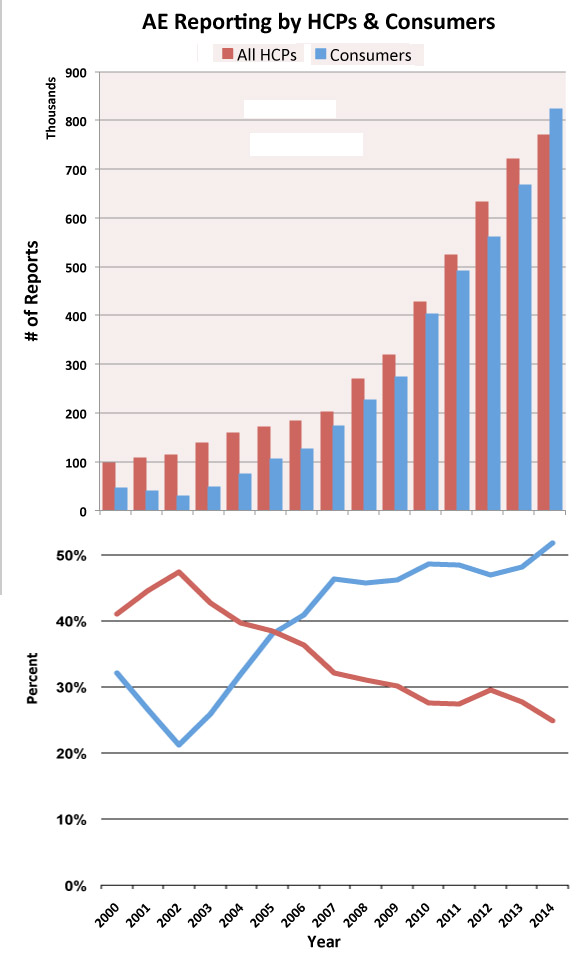
Not only is the FDA receiving more AERs every year, the number of serious AEs and AEs involving death are also increasing rather dramatically as shown in the following chart.
 |
| Click on image for an enlarged view. |
The Harvard Edmond J. Safra Center for Ethics reports that a forthcoming article in the Journal of Law, Medicine and Ethics (“Institutional Corruption of Pharmaceuticals and the Myth of Safe and Effective Drugs“) will present “systematic, quantitative evidence that since the industry started making large contributions to the FDA for reviewing its drugs, as it makes large contributions to Congressmen who have promoted this substitution for publicly funded regulation, the FDA has sped up the review process with the result that drugs approved are significantly more likely to cause serious harm, hospitalizations, and deaths. New FDA policies are likely to increase the epidemic of harms” (read “Risky Drugs: Why The FDA Cannot Be Trusted“).
The authors of the JLME paper contend that “in return for paying user fees, companies required the FDA to guarantee that it would review priority applications within six months and standard applications within 12 months of submission. Shortened review times led to substantial increases in serious harms.” I thought this was an interesting because I have been following the negative correlation between the rise in user fees and the decrease in FDA warning letters, which may encourage approval of dangerous drugs by the FDA (see the data charted in this post: “Do PDUFA Fees Encourage Approval of Dangerous Drugs and/or Undermine FDA’s Regulatory Oversight of Approved Drugs?“).
So, I decided to plot the number of AERs involving serious side effects along with the rise in user fees collected by the FDA from pharma companies and came up with the following chart.
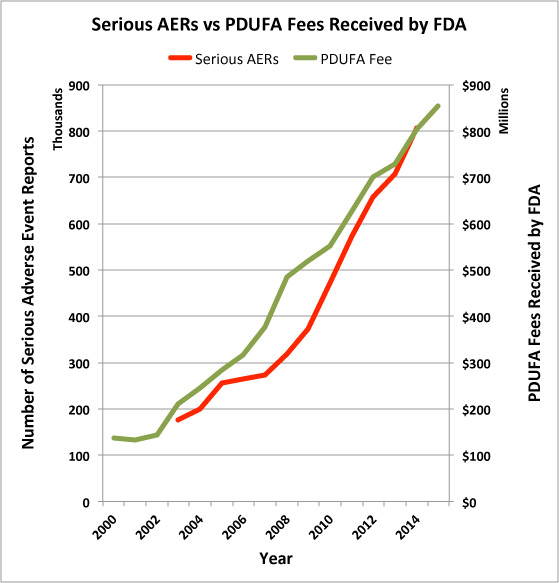
Of course, this does not prove a correlation between PDUFA fees and serious AERs, but it does lend support to the “institutional corruption” argument posit in the above article.








")



{kind=link}